Chemistry Journal of Moldova

- Journal Citation Reports for year 2025:
- Impact Factor (IF) 0.5;
- 5-year Journal IF 0.6;
- SCOPUS Journal Metrics for year 2025:
- CiteScore 0.9;
- Source Normalized Impact per Paper (SNIP) 0.334;
- SCImago Journal Rank (SJR) 0.152.



TOP DOWNLOADED ARTICLES
1. SONOCHEMICAL SYNTHESIS OF HEMATITE
NANOPARTICLES
Mihail Iacob
(downloads: 1328)
2. THE ROLE OF HALOGEN BONDING IN THE
SYNTHESIS AND DESIGN OF TAP COMPLEXES
Namiq Shikhaliyev, Ayten Niyazova, Abel Maharramov, Agaisa Askerov, Nazrin Zeynalli, Viktor Khrustalev, Peri Huseynova
(downloads: 901)
3. SILVER AND ZINC NANOPARTICLES
BIOSYNTHESIS USING LAUREL EXTRACT AND
INVESTIGATION OF THE PHOTOCATALYTIC
PROPERTIES
Recep Taş, Ebru Köroğlu, Ahmet Karakuş, Ali Savaş Bülbül, Nilay Akkuş Taş
(downloads: 900)
4. SYNTHESIS OF DIFFERENT STRUCTURAL TYPES
OF ZEOLITES IN THE
HALLOYSITE-DOLOMITE-OBSIDIAN SYSTEM
Gunel Mamedova
(downloads: 859)
5. DERIVATIZATION TECHNIQUES BASED ON
CHARGE TRANSFER REACTIONS FOR
SPECTROPHOTOMETRIC DETERMINATION OF
JOSAMYCIN IN VARIOUS DOSAGE FORMS
Abdelghani Mahmoudi and Ann Van Schepdael
(downloads: 831)
6. A NOVEL GREEN SYNTHESIS OF NICKEL OXIDE
NANOPARTICLES USING ARABIC GUM
Saeid Taghavi Fardood, Ali Ramazani, Sajjad Moradi
(downloads: 803)
7. SYNTHESIS, CHARACTERIZATION AND
MOLECULAR DOCKING OF CHLORO-SUBSTITUTED
HYDROXYXANTHONE DERIVATIVES
Emmy Yuanita, Harno Dwi Pranowo, Mustofa Mustofa, Respati Tri Swasono, Jufrizal Syahri, Jumina Jumina
(downloads: 644)
8. ROLE OF CYCLODEXTRINS IN NEW
ANTIMYCOBACTERIAL FORMULATIONS
Veaceslav Boldescu, Fliur Macaev, Gheorghe Duca
(downloads: 499)
9. ORGANOCHLORINE PESTICIDES RESIDUES IN
SOIL OF SOROCA DISTRICT, REPUBLIC OF
MOLDOVA
Elena Culighin
(downloads: 479)
10. INVESTIGATION OF VARIOUS INFLUENCING
FACTORS OF HYDROTHERMAL SYNTHESIS OF
ANALCIME ZEOLITE
Gunel Mamedova, Gunel Nasirli
(downloads: 473)
NANOPARTICLES
Mihail Iacob
(downloads: 1328)
2. THE ROLE OF HALOGEN BONDING IN THE
SYNTHESIS AND DESIGN OF TAP COMPLEXES
Namiq Shikhaliyev, Ayten Niyazova, Abel Maharramov, Agaisa Askerov, Nazrin Zeynalli, Viktor Khrustalev, Peri Huseynova
(downloads: 901)
3. SILVER AND ZINC NANOPARTICLES
BIOSYNTHESIS USING LAUREL EXTRACT AND
INVESTIGATION OF THE PHOTOCATALYTIC
PROPERTIES
Recep Taş, Ebru Köroğlu, Ahmet Karakuş, Ali Savaş Bülbül, Nilay Akkuş Taş
(downloads: 900)
4. SYNTHESIS OF DIFFERENT STRUCTURAL TYPES
OF ZEOLITES IN THE
HALLOYSITE-DOLOMITE-OBSIDIAN SYSTEM
Gunel Mamedova
(downloads: 859)
5. DERIVATIZATION TECHNIQUES BASED ON
CHARGE TRANSFER REACTIONS FOR
SPECTROPHOTOMETRIC DETERMINATION OF
JOSAMYCIN IN VARIOUS DOSAGE FORMS
Abdelghani Mahmoudi and Ann Van Schepdael
(downloads: 831)
6. A NOVEL GREEN SYNTHESIS OF NICKEL OXIDE
NANOPARTICLES USING ARABIC GUM
Saeid Taghavi Fardood, Ali Ramazani, Sajjad Moradi
(downloads: 803)
7. SYNTHESIS, CHARACTERIZATION AND
MOLECULAR DOCKING OF CHLORO-SUBSTITUTED
HYDROXYXANTHONE DERIVATIVES
Emmy Yuanita, Harno Dwi Pranowo, Mustofa Mustofa, Respati Tri Swasono, Jufrizal Syahri, Jumina Jumina
(downloads: 644)
8. ROLE OF CYCLODEXTRINS IN NEW
ANTIMYCOBACTERIAL FORMULATIONS
Veaceslav Boldescu, Fliur Macaev, Gheorghe Duca
(downloads: 499)
9. ORGANOCHLORINE PESTICIDES RESIDUES IN
SOIL OF SOROCA DISTRICT, REPUBLIC OF
MOLDOVA
Elena Culighin
(downloads: 479)
10. INVESTIGATION OF VARIOUS INFLUENCING
FACTORS OF HYDROTHERMAL SYNTHESIS OF
ANALCIME ZEOLITE
Gunel Mamedova, Gunel Nasirli
(downloads: 473)


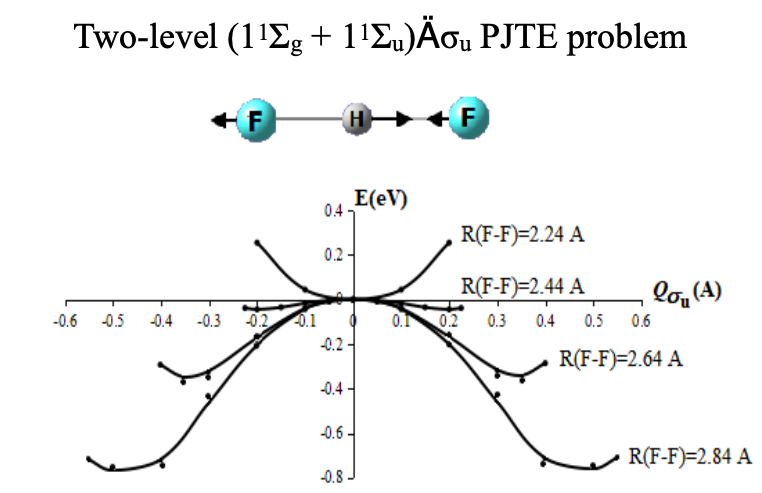
PSEUDO JAHN-TELLER ORIGIN OF THE PROTON-TRANSFER ENERGY BARRIER IN THE HYDROGEN-BONDED [FHF]- SYSTEM
Author(s):
Field: Physical chemistry and chemical physics
Type: Research paper
Issue: 2021 Volume 16, no.1
Pages: 115-120
Natalia Gorinchoy, Iolanta Balan, Victor Polinger, Isaak Bersuker
Field: Physical chemistry and chemical physics
Type: Research paper
Issue: 2021 Volume 16, no.1
Pages: 115-120
proton transfer, hydrogen bond, pseudo Jahn–Teller effect, potential energy surface, bifluoride anion.
Full Text (PDF): Download
Graphical Abstract: The results of ab initio calculations of the adiabatic potential energy surfaces for the proton-bound [FHF]- system at different F-F distances have been rationalized in the framework of the vibronic theory. It is shown that the instability of the symmetric D∞h structure at increased F∙∙∙F distances and the proton displacement to one of the fluorine atoms is due to the pseudo Jahn–Teller mixing of the ground 1Σg electronic state with the lowest excited state of 1Σu symmetry through the asymmetric σu vibrational mode.
Downloads: 138